Main output matrices
Table of Contents
This section describes where are the main connectomes generated by the pipeline, and how you can visualize them using python or R.
All the outputs are BIDS conform and stored under their correspondent directory (e.g. mpc, func, dwi, dist).
This example will use the subject HC001, session 01 from the MICs dataset, and all paths will be relative to the subject directory or out/micapipe/sub-HC001_ses01/
Download code examples: matrices
Python Jupyter notebook: 'tutorial_main_output_matrices.ipynb'
Organization of the outputs
If you are inside the out directory, where the pipeline’s outputs are stored, two folders can be found inside, one for each pipeline used.
For this dataset, the main structure should look like this:
out
├── fastsurfer
└── micapipe_v0.2.0
Inside the directory called micapipe_v0.2.0 a new directory will be created for each subject processed, defined with the string sub- and the subject’s identification. Three more files can be found:
dataset_description.json: This file contains the last version of micapipe that was used and, the inherited information about the dataset used.
micapipe_processed_sub.csv: This is a csv table stores information about the modules processed by subject, date of last run and the status (complete or incomplete).
micapipe_group-QC.pdf: This file is generated with the option-QC, it is a pdf that eases the visualization of the processed subjects.
micapipe_v0.2.0/
├── micapipe_processed_sub.csv
├── dataset_description.json
├── micapipe_group-QC.pdf
├── sub-HC001
├── sub-HC002
├── sub-HC003
...
Inside each subject’s directory you’ll find the session folders, unless you have a single session and you don’t specify the ses- in your dataset.
Six main directories are inside each subject folder: anat, dist, dwi, func, logs, maps, mpc, parc, QC and xfm. The connectomes are stored in four main directories:
Geodesic distance connectome:
distMicrostructural profile connectome:
mpc/<acquisition>Structural connectome (DWI):
dwi/connectomesFunctional connectome (fMRI):
func/<acquisition>/surf
The structure of the subject HC001 directories is shown below:
sub-HC001/
└── ses-01
├── anat
├── dist
├── dwi
│ ├── connectomes # DWI connectomes
│ └── eddy # fsl eddy outputs
├── func
│ ├── desc-me_task-rest_bold
│ │ ├── surf
│ │ └── volumetric
│ └── desc-me_task-semantic_bold
│ ├── surf
│ └── volumetric
├── logs # log files
├── maps
├── mpc
├── QC
│ └── eddy_QC # fsl eddy_quad outputs
├── surf
└── xfm # Transformation matrices and warpfields
In the following examples, we’ll focus on how to load and visualize the connectome matrices of a single subject.
Even though we generate up to 18 connectomes, we’ll only use the atlas schaefer-400 for visualization and practicality purposes.
All the paths are relative to the subject’s directory, which in our case is out/micapipe_v0.2.0/sub-HC001/ses-01.
This example uses the packages nilearn, nibabel, numpy and matplotlib for python, and gifti, RColorBrewer and viridis for R.
The first step in both languages is to set the environment:
# Load required packages
import os
import numpy as np
import nibabel as nib
from nilearn import plotting
import matplotlib as plt
# Set the working directory to the 'out' directory
os.chdir("/data_/mica3/BIDS_MICs/derivatives") # <<<<<<<<<<<< CHANGE THIS PATH TO YOUR OUT DIRECTORY
# This variable will be different for each subject
sub='HC001' # <<<<<<<<<<<< CHANGE THIS SUBJECT's ID
ses='01' # <<<<<<<<<<<< CHANGE THIS SESSION
subjectID=f'sub-{sub}_ses-{ses}'
subjectDir=f'micapipe_v0.2.0/sub-{sub}/ses-{ses}'
# Here we define the atlas
atlas='schaefer-400'
# Set the environment
require("RColorBrewer")
require("viridis")
require("gifti")
# Set the working directory to your subject's directory
setwd("out/micapipe_v0.2.0/sub-HC001/ses-01")
# This variable will be different for each subject
subjectID <- 'sub-HC001_ses-01'
# Here we define the atlas
atlas <- 'schaefer-400'
Structural connectome
- Structural connectomes are stored in the
dwi/connectomesdirectory. One main connectomes is generated per atlas, and are identified with a specific string: full-connectome: Full connectome has cerebellar, subcortical and cortical nodes.
Additionally, the edge length of the previous connectomes is stored in a different file with the string edgeLengths.
Two files per atlas are generated by the pipeline, the main organization is shown below:
dwi/connectomes/
├── sub-HC005_ses-01_space-dwi_atlas-schaefer-400_desc-iFOD2-40M-SIFT2_full-connectome.shape.gii
└── sub-HC005_ses-01_space-dwi_atlas-schaefer-400_desc-iFOD2-40M-SIFT2_full-edgeLengths.shape.gii
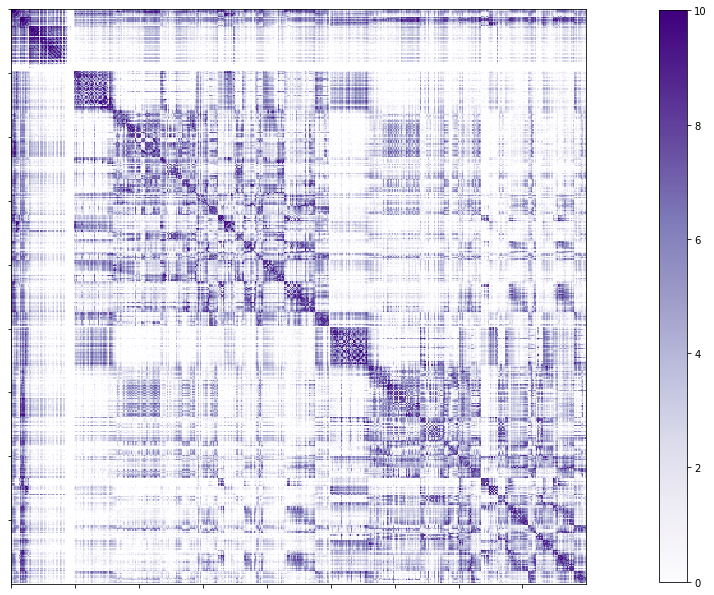
Full structural connectome
# Set the path to the the structural cortical connectome
cnt_sc_cor = f'{subjectDir}/dwi/connectomes/{subjectID}_space-dwi_atlas-{atlas}_desc-iFOD2-40M-SIFT2_full-connectome.shape.gii'
# Load the cortical connectome
mtx_sc = nib.load(cnt_sc_cor).darrays[0].data
# Fill the lower triangle of the matrix
mtx_scSym = np.triu(mtx_sc,1)+mtx_sc.T
# Plot the log matrix
corr_plot = plotting.plot_matrix(np.log(mtx_scSym), figure=(10, 10), labels=None, cmap='Purples', vmin=0, vmax=10)
# Set the path to the the structural cortical connectome
cnt_sc_cor <- paste0('dwi/connectomes/', subjectID, '_space-dwi_atlas-', atlas, '_desc-iFOD2-40M-SIFT2_full-connectome.shape.gii')
# Load the cortical connectome
mtx_sc <- readgii(cnt_sc_cor)$data$shape
# Fill the lower triangle of the matrix
mtx_sc[lower.tri(mtx_sc)] <- t(mtx_sc)[lower.tri(mtx_sc)]
# Plot the log matrix
image(log(mtx_sc), axes=FALSE, main=paste0("SC ", atlas), col=brewer.pal(9, "Purples"))
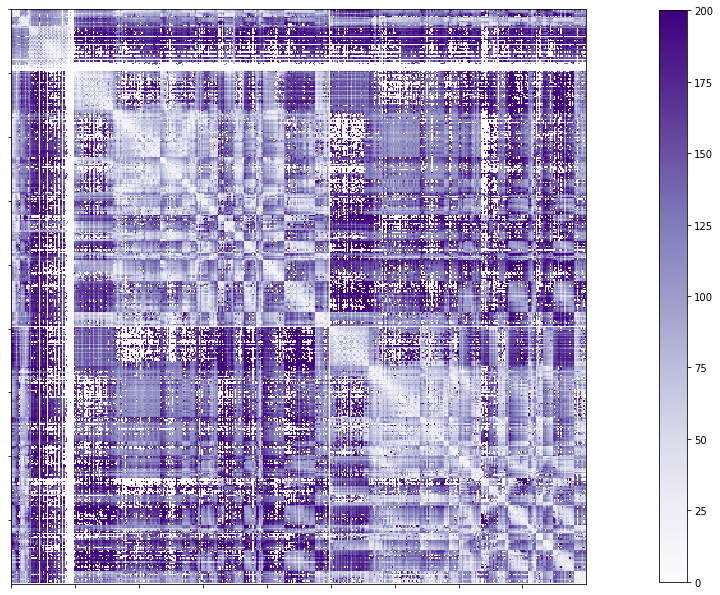
Full structural connectome edge lengths
# Set the path to the the structural cortical connectome
cnt_sc_EL = cnt_sc_cor= f'{subjectDir}/dwi/connectomes/{subjectID}_space-dwi_atlas-{atlas}_desc-iFOD2-40M-SIFT2_full-edgeLengths.shape.gii'
# Load the cortical connectome
mtx_scEL = nib.load(cnt_sc_EL).darrays[0].data
# Fill the lower triangle of the matrix
mtx_scELSym = np.triu(mtx_scEL,1)+mtx_scEL.T
# Plot the log matrix
corr_plot = plotting.plot_matrix(mtx_scELSym, figure=(10, 10), labels=None, cmap='Purples', vmin=0, vmax=200)
# Set the path to the the structural cortical connectome
cnt_sc_EL <- paste0('dwi/connectomes/', subjectID, '_space-dwi_atlas-', atlas, '_desc-iFOD2-40M-SIFT2_full-edgeLengths.shape.gii')
# Load the cortical connectome
mtx_scEL <- readgii(cnt_sc_EL)$data$shape
# mtx_scEL <- as.matrix(read.csv(cnt_sc_EL, sep=" ", header=FALSE,))
# Fill the lower triangle of the matrix
mtx_scEL[lower.tri(mtx_scEL)] <- t(mtx_scEL)[lower.tri(mtx_scEL)]
# Plot the log matrix
image(log(mtx_scEL), axes=FALSE, main=paste0("SC ", atlas), col=brewer.pal(9, "Purples"))
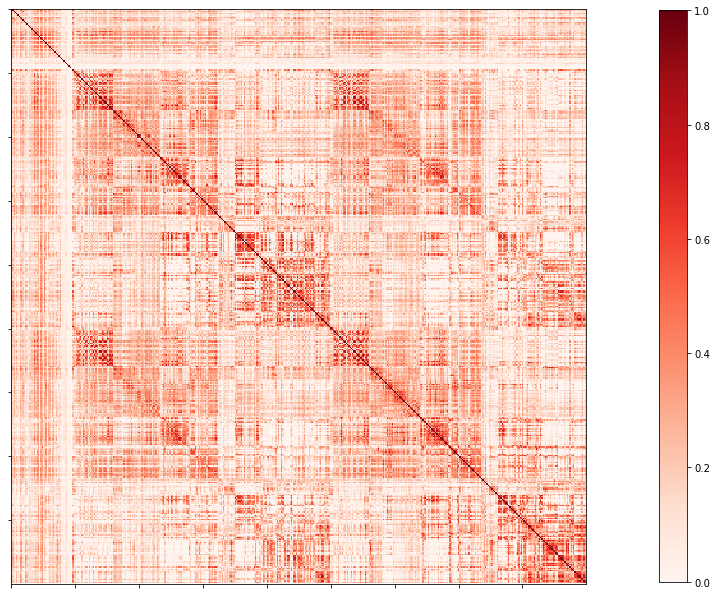
Functional connectome
For each atlas, one file is generated: the functional connectome (desc-FC.shape.gii) and
The time-series of that atlas is only stored in the surface fsLR-32k (surf-fsLR-32k_desc-timeseries_clean.shape.gii).
func/<acquisition>/surf/
└── sub-HC005_ses-01_surf-fsLR-32k_atlas-schaefer-400_desc-FC.shape.gii
# Set the path to the the functional connectome
# acquisitions
func_acq='desc-se_task-rest_acq-AP_bold'
cnt_fs = subjectDir + f'/func/{func_acq}/surf/{subjectID}_surf-fsLR-32k_atlas-{atlas}_desc-FC.shape.gii'
# Load the cortical connectome
mtx_fs = nib.load(cnt_fs).darrays[0].data
# Fill the lower triangle of the matrix
mtx_fcSym = np.triu(mtx_fs,1)+mtx_fs.T
# Plot the matrix
corr_plot = plotting.plot_matrix(mtx_fcSym, figure=(10, 10), labels=None, cmap='Reds', vmin=0, vmax=1)
# Set the path to the the functional connectome
acq_func <- 'desc-se_task-rest_acq-AP_bold'
cnt_fs <- paste0('func/', acq_func, '/surf/', subjectID, '_surf-fsLR-32k_atlas-', atlas, '_desc-FC.shape.gii')
# Load the cortical connectome
mtx_fs <- readgii(cnt_fs)$data$shape
# mtx_fs <- as.matrix(read.csv(cnt_fs, sep=" ", header=FALSE))
# Fill the lower triangle of the matrix
mtx_fs[lower.tri(mtx_fs)] <- t(mtx_fs)[lower.tri(mtx_fs)]
# Plot the matrix
image(mtx_fs, axes=FALSE, main=paste0("FC ", atlas), col=brewer.pal(9, "Reds"))
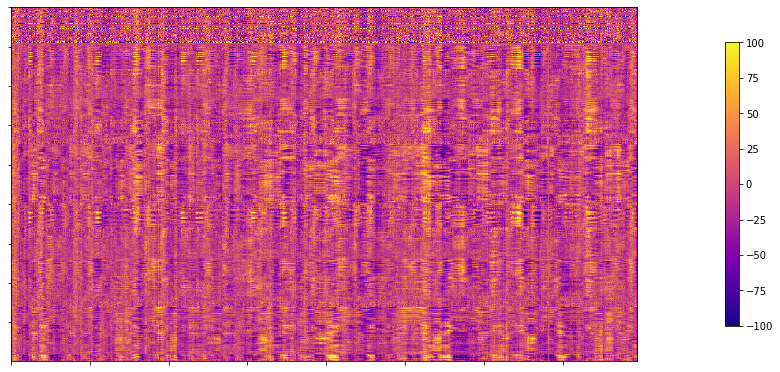
Resting state time series
# Set the path to the the time series file
cnt_time = subjectDir + f'/func/{func_acq}/surf/{subjectID}_surf-fsLR-32k_desc-timeseries_clean.shape.gii'
# Load the time series
mtx_time = nib.load(cnt_time).darrays[0].data
# Plot as a matrix
corr_plot = plotting.plot_matrix(mtx_time, figure=(300, 10), labels=None, cmap='plasma', vmin=-100, vmax=100)
# Set the path to the the time series file
cnt_time <- paste0('func/', acq_func, '/surf/', subjectID, '_surf-fsLR-32k_desc-timeseries_clean.shape.gii')
# Load the time series
mtx_time <- readgii(cnt_time)$data$shape
# mtx_time <- as.matrix(read.csv(cnt_time, sep=" ", header=FALSE))
# Plot as a matrix
image(mtx_time, axes=FALSE, main=paste0("Time series ", atlas), col=plasma(64))
MPC connectome
For each atlas, two files are generated: the microstructural profile covariance connectome (desc-MPC.shape.gii) and the intensity profile of that atlas (desc-intensity_profiles.shape.gii).
mpc/<acquisition>/
├── sub-HC005_ses-01_atlas-schaefer-400_desc-intensity_profiles.shape.gii
└── sub-HC005_ses-01_atlas-schaefer-400_desc-MPC.shape.gii
# Set the path to the the MPC cortical connectome
mpc_acq='acq-T1map'
ccnt_mpc = subjectDir + f'/mpc/{mpc_acq}/{subjectID}_atlas-{atlas}_desc-MPC.shape.gii'
# Load the cortical connectome
mtx_mpc = nib.load(cnt_mpc).darrays[0].data
# Fill the lower triangle of the matrix
mtx_mpcSym = np.triu(mtx_mpc,1)+mtx_mpc.T
# Plot the matrix
corr_plot = plotting.plot_matrix(mtx_mpcSym, figure=(10, 10), labels=None, cmap='Greens')
# Set the path to the the MPC cortical connectome
cnt_mpc <- paste0('mpc/acq-T1map/', subjectID, '_atlas-', atlas, '_desc-MPC.shape.gii')
# Load the cortical connectome
mtx_mpc <- readgii(cnt_mpc)$data$shape
# Fill the lower triangle of the matrix
mtx_mpc[lower.tri(mtx_mpc)] <- t(mtx_mpc)[lower.tri(mtx_mpc)]
# Plot the matrix
image(mtx_mpc, axes=FALSE, main=paste0("MPC ", atlas), col=brewer.pal(9, "Greens"))

Intensity profiles
# Set the path to the Intensity profiles file
cnt_int = subjectDir + f'/mpc/{mpc_acq}/{subjectID}_atlas-{atlas}_desc-intensity_profiles.shape.gii'
# Load the Intensity profiles
mtx_int = nib.load(cnt_int).darrays[0].data
# Plot as a matrix
corr_plot = plotting.plot_matrix(mtx_int, figure=(20,10), labels=None, cmap='Greens', colorbar=False)
# Set the path to the Intensity profiles file
cnt_int <- paste0('mpc/acq-T1map/', subjectID, '_atlas-', atlas, '_desc-intensity_profiles.shape.gii')
# Load the time series
mtx_int <- readgii(cnt_int)$data$shape
#mtx_int <- as.matrix(read.csv(cnt_int, sep=" ", header=FALSE))
# Plot as a matrix
image(t(mtx_int), axes=FALSE, main=paste0("Intensity profiles", atlas), col=brewer.pal(9, "Greens"))

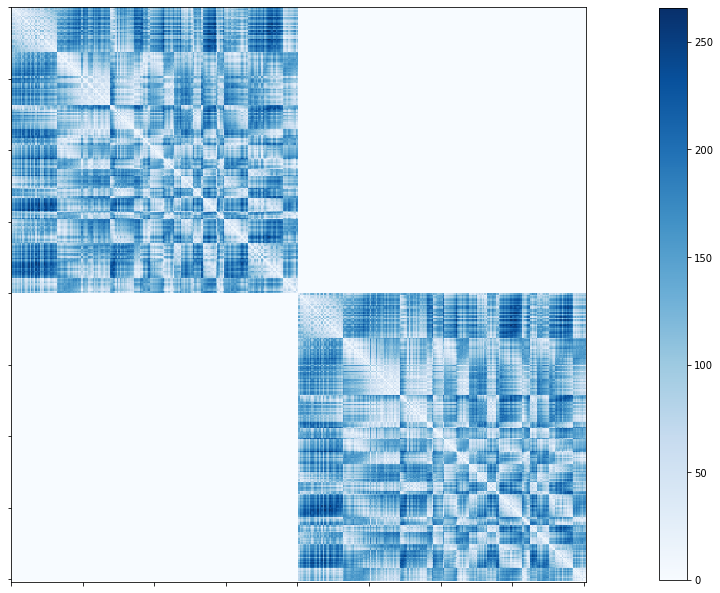
Geodesic distance connectome
Only one file per atlas is generated by this module:
dist/
└── sub-HC005_ses-01_atlas-schaefer-400_GD.shape.gii
# Set the path to the the geodesic distance connectome
cnt_gd = f'{subjectDir}/dist/{subjectID}_atlas-{atlas}_GD.shape.gii'
# Load the cortical connectome
mtx_gd = nib.load(cnt_gd).darrays[0].data
# Plot the matrix
corr_plot = plotting.plot_matrix(mtx_gd, figure=(10, 10), labels=None, cmap='Blues')
# Set the path to the the geodesic distance connectome
cnt_gd <- paste0('dist/', subjectID, '_atlas-', atlas, '_GD.shape.gii')
# Load the cortical connectome
mtx_gd <- readgii(cnt_gd)$data$shape
# Plot the matrix
image(t(mtx_gd), axes=FALSE, main=paste0("GD ", atlas), col=brewer.pal(9, "Blues"))